
I. Co to jest Fenyloketonuria?
A. Definicja choroby

Fenyloketonuria (PKU) jest rzadką dziedziczną chorobą genetyczną, która powoduje zaburzenie metabolizmu aminokwasu fenyloalaniny. Osoby cierpiące na PKU nie mogą prawidłowo przetwarzać fenyloalaniny, co prowadzi do jej gromadzenia się we krwi i powoduje uszkodzenie mózgu.
B. Historia odkrycia PKU
PKU zostało odkryte przez norweskiego lekarza Asbjørna Føllinga w 1934 roku. Przypadkowo odkrył obecność dużych ilości fenyloalaniny we mleku pacjenta z zaburzeniami neurologicznymi. To odkrycie doprowadziło do zrozumienia związku między fenyloalaniną a PKU.
C. Znaczenie wczesnej diagnozy
Wczesna diagnoza PKU jest kluczowa dla zapobiegania poważnym konsekwencjom tej choroby. Dzięki wcześniejszemu wykryciu, pacjenci mogą być wprowadzeni na specjalną dietę niskobiałkową, która pozwala uniknąć uszkodzeń mózgu i innych powikłań wynikających z nadmiernego gromadzenia się fenyloalaniny. Dlatego regularne badania przesiewowe są istotne dla zapewnienia szybkiej diagnozy i skutecznego leczenia PKU.
II. Genetyczne podstawy Fenyloketonurii
A. Mutacje w genie PAH
Fenyloketonuria jest spowodowana mutacjami w genie PAH, który zawiera instrukcje dla produkcji enzymu fenylalaninohydrolazy (PH). Enzym ten jest kluczowy dla prawidłowego metabolizmu fenyloalaniny. Mutacje w genie PAH prowadzą do niedoboru lub braku funkcji tego enzymu, co skutkuje gromadzeniem się fenyloalaniny we krwi i tkankach.
B. Dziedziczenie autosomalne recesywne
Fenyloketonuria jest dziedziczona w sposób autosomalny recesywny, co oznacza, że aby osoba wykazywała objawy tej choroby, musi odziedziczyć mutowany gen od obydwu rodziców. Osoby z jednym zmutowanym allelem tego genu są nosicielami, ale nie wykazują objawów PKU. Dziedziczenie recesywne wpływa na to, że choroba może pojawić się nawet jeśli nie występują objawy u rodziców, co sprawia, że PKU często jest niespodziewaną diagnozą urodzeniową.
III. Mechanizm działania: Metabolizm fenyloalaniny
A. Rola fenyloalaniny w organizmie
Fenyloalanina jest aminokwasem, który pochodzi z pożywienia i jest niezbędny dla organizmu do produkcji białek i innych związków. Jednakże, u osób z fenyloketonurią, fenyloalanina nie jest efektywnie metabolizowana, co prowadzi do jej nadmiernego gromadzenia się we krwi i tkankach.
B. Brak aktywności enzymu hydroksylazy fenyloalaninowej
W organizmach osób z fenyloketonurią występuje brak aktywności enzymu hydroksylazy fenyloalaninowej, który jest niezbędny do przekształcenia fenyloalaniny w tyrozynę. Bez tego enzymu fenyloalanina pozostaje niewykorzystana i gromadzi się, co może prowadzić do poważnych skutków zdrowotnych.
C. Skutki nadmiaru fenyloalaniny
Nadmiar fenyloalaniny we krwi i tkankach osób z fenyloketonurią może prowadzić do uszkodzenia mózgu, co skutkuje trwałymi deficytami intelektualnymi i neurologicznymi. Dlatego kluczowym elementem leczenia PKU jest utrzymanie niskich poziomów fenyloalaniny poprzez dietę i monitorowanie, aby minimalizować ryzyko powikłań zdrowotnych związanych z nadmiernym gromadzeniem się tego aminokwasu.
IV. Objawy Fenyloketonurii
A. Wczesne objawy u niemowląt
- Charakterystyczny zapach moczu: Niemowlęta z fenyloketonurią często mają charakterystyczny, myszawy zapach moczu, który jest spowodowany nadmiernym wydalaniem substancji chemicznych związanych z nadmiarem fenyloalaniny.
- Opóźnienia w rozwoju: Osoby z fenyloketonurią mogą doświadczać opóźnień w osiąganiu kolejnych między etapów rozwojowych, takich jak zdolność chodzenia, mówienia czy nabywania umiejętności społecznych.
B. Objawy neurologiczne
- Upośledzenie umysłowe: Nadmiar fenyloalaniny może prowadzić do uszkodzenia mózgu i związanej z tym trwałej obniżonej sprawności intelektualnej.
- Problemy z mową i zachowaniem: Osoby z fenyloketonurią mogą mieć trudności z normalnym rozwojem mowy oraz mogą wykazywać nieprawidłowości w zachowaniu, takie jak hiperaktywność czy trudności w koncentracji.
C. Objawy fizyczne
- Zaburzenia wzrostu i budowy ciała: Nadmiar fenyloalaniny może wpływać na wzrost i rozwój fizyczny, prowadząc do opóźnień w rozwoju ciała oraz zaburzeń w budowie anatomicznej.
- Problemy skórne: W przypadku fenyloketonurii mogą się pojawić różne problemy skórne, takie jak zmiany pigmentacyjne, trądzik czy suchość skóry, będące wynikiem zaburzeń metabolicznych związanych z nadmiarem fenyloalaniny we krwi.
V. Diagnostyka Fenyloketonurii
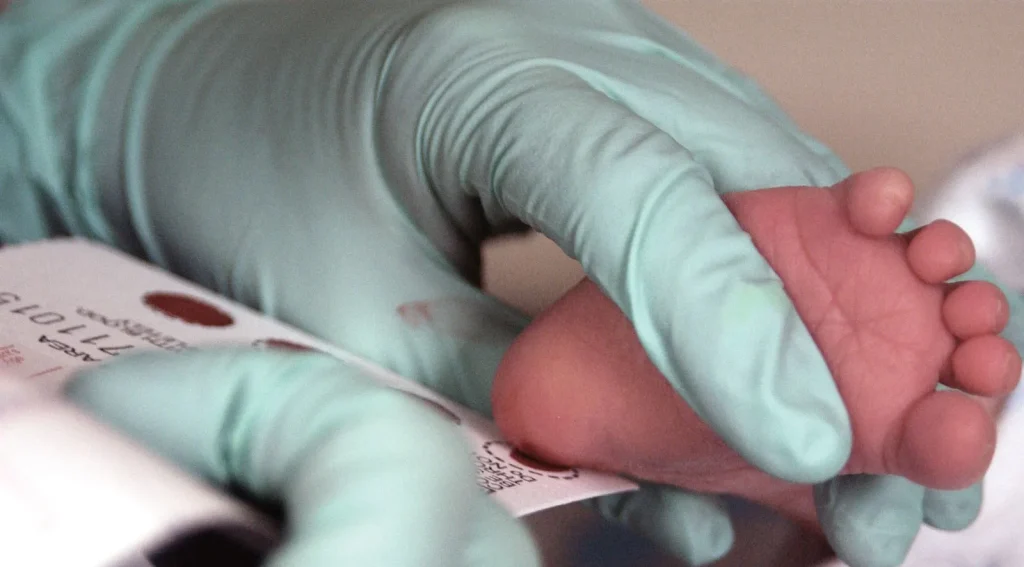
A. Testy przesiewowe noworodków
- Badanie Guthriego: Jest to standardowy test przesiewowy stosowany u noworodków, polegający na pobraniu kropli krwi z pięty dziecka w celu zbadania poziomu fenyloalaniny. Wzrost poziomu fenyloalaniny może sugerować obecność fenyloketonurii.
- Testy tandemowej spektrometrii mas: Jest to zaawansowana technika laboratoryjna, która umożliwia dokładne pomiaranie stężenia różnych związków zawartych we krwi, w tym fenyloalaniny. Jest to skuteczna metoda diagnostyczna przy podejrzeniu fenyloketonurii.
B. Diagnostyka prenatalna
- Analiza DNA: Wykorzystuje się testy genetyczne w celu zidentyfikowania mutacji genetycznych związanych z fenyloketonurią. Diagnostyka prenatalna pozwala na wczesne wykrycie choroby u płodu.
- Biopsja kosmówki: Jest to procedura diagnostyczna, podczas której pobierana jest próbka tkanki kosmówki płodu do analizy genetycznej w celu oceny ryzyka wystąpienia fenyloketonurii.
C. Potwierdzenie diagnozy
- Poziom fenyloalaniny we krwi: W celu potwierdzenia diagnozy fenyloketonurii, wykonywany jest pomiar stężenia fenyloalaniny we krwi. Podwyższony poziom fenyloalaniny może być potwierdzeniem obecności choroby.
- Dodatkowe testy biochemiczne: Do potwierdzenia diagnozy fenyloketonurii mogą być wykonywane dodatkowe testy biochemiczne, które mogą obejmować badanie stężenia innych substancji we krwi lub moczem, aby potwierdzić zaburzenia metaboliczne charakterystyczne dla choroby.
VI. Dieta niskofenyloalaninowa
A. Podstawy diety
- Dieta niskofenyloalaninowa jest niezbędna dla osób cierpiących na fenyloketonurię, gdyż nie są one w stanie prawidłowo metabolizować fenyloalaniny, co może prowadzić do poważnych konsekwencji zdrowotnych, włącznie z uszkodzeniem mózgu.
- Podstawowym celem diety jest ograniczenie spożycia fenyloalaniny poprzez eliminację lub ograniczenie produktów bogatych w ten aminokwas.
B. Lista dozwolonych i zakazanych produktów
- Produkty dozwolone: Osoby z fenyloketonurią mogą spożywać produkty z niską zawartością fenyloalaniny, takie jak:
- Warzywa z wyjątkiem rzepaku, pomidorów, cukinii oraz groszku;
- Owoce: jabłka, gruszki, banany, brzoskwinie, śliwki;
- Ryby i mięsa chude;
- Mleko i produkty mleczne o niskiej zawartości białka;
- Produkty zbożowe niskobiałkowe, takie jak ryż, ziemniaki, kasza gryczana;
- Produkty słodzące niskofenyloalaninowe.
- Produkty zakazane: Osoby z fenyloketonurią powinny unikać produktów zawierających większe ilości fenyloalaniny, w tym:
- Mięso, ryby i warzywa wędzone;
- Wędliny;
- Produkty mleczne pełne tłuszczu;
- Orzechy, nasiona;
- Czekolada, kakao, słodycze zawierające fenyloalaninę;
- Produkty zawierające aspartam (słodzik zawierający fenyloalaninę).
Przestrzeganie diety niskofenyloalaninowej jest kluczowe dla osób cierpiących na fenyloketonurię, gdyż pozwala uniknąć toksycznych skutków nadmiaru fenyloalaniny w organizmie. W przypadku wątpliwości co do składu diety, zaleca się konsultację z dietetykiem specjalizującym się w dietach terapeutycznych dla osób z fenyloketonurią.
VII. Suplementacja i substytuty białkowe
A. Preparaty aminokwasowe
- Osoby cierpiące na fenyloketonurię często potrzebują suplementacji aminokwasów, zwłaszcza fenyloalaniny, aby zapewnić odpowiednie działanie organizmu. Preparaty aminokwasowe są tworzone specjalnie dla osób z fenyloketonurią i zawierają kontrolowaną ilość fenyloalaniny, co pozwala na uzupełnienie niedoborów bez ryzyka toksycznego nagromadzenia tego aminokwasu.
- Preparaty aminokwasowe są zazwyczaj dostosowywane indywidualnie do potrzeb pacjenta i stosowane pod ścisłą kontrolą lekarza specjalizującego się w leczeniu fenyloketonurii.
B. Witaminy i minerały
- Osoby z fenyloketonurią często potrzebują uzupełnienia witamin i minerałów, zwłaszcza tych, których naturalne źródła są ograniczone w diecie.
- Niektóre witaminy i minerały, takie jak witamina B12, żelazo, wapń i witamina D, mogą być niedoborowe ze względu na obostrzenia żywieniowe związane z fenyloketonurią. Konieczna jest zatem regularna kontrola poziomu tych substancji i ewentualna suplementacja pod nadzorem lekarza.
Suplementacja i stosowanie substytutów białkowych są kluczowe dla utrzymania zdrowia i prawidłowego rozwoju u osób cierpiących na fenyloketonurię. Wszelkie suplementy i substytuty białkowe powinny być stosowane zgodnie z zaleceniami lekarza oraz dietetyka specjalizującego się w fenyloketonurii, aby zapewnić prawidłową równowagę aminokwasową i składników odżywczych w organizmie.
VIII. Nowoczesne metody leczenia
A. Terapia enzymatyczna (Pegvaliase)
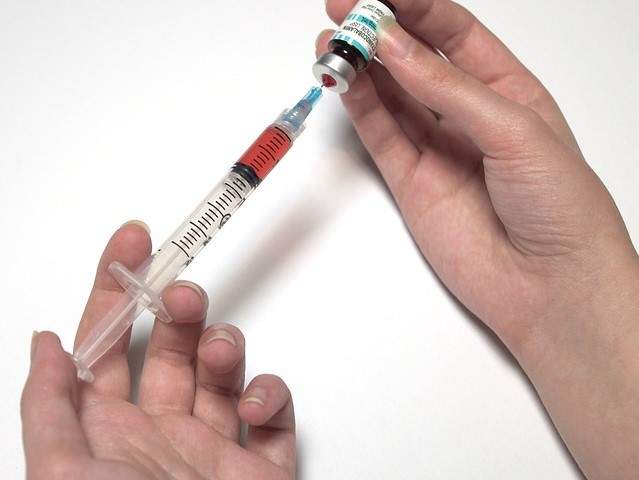
- Terapia enzymatyczna, z wykorzystaniem preparatu Pegvaliase (handlowa nazwa Palynziq), jest jedną z nowoczesnych metod leczenia fenyloketonurii. Pegvaliase jest rekombinowanym enzymem (fenylalaniną amoniowującym enzymem) iniekcjami podskórnymi, który pomaga w metabolizowaniu nadmiaru fenyloalaniny w organizmie, zmniejszając poziom tego aminokwasu do bezpiecznych wartości.
- Terapia enzymatyczna przy użyciu Pegvaliase może być stosowana u pacjentów, u których tradycyjne metody leczenia, takie jak dietę niskobiałkową, okazały się nieskuteczne lub trudne do kontrolowania. Jest to obiecujące podejście w leczeniu fenyloketonurii i może poprawić jakość życia pacjentów.
B. Terapie genowe: obiecujące badania
- Terapie genowe to innowacyjne podejście w leczeniu fenyloketonurii, które wciąż jest w fazie badań klinicznych. Polega to na stosowaniu technik genetycznych do naprawy defektu genetycznego odpowiedzialnego za fenyloketonurię. Poprzez wprowadzenie zdrowego genu lub zmodyfikowanie defektywnego genu poprzez terapię genową, naukowcy starają się zapewnić pacjentom zdolność do właściwej metabolizacji fenyloalaniny.
- Badania nad terapiami genowymi w fenyloketonurii są nadal w fazie eksperymentalnej, ale stanowią obiecujące rozwiązanie dla osób z tą chorobą genetyczną. Monitorowanie postępów w dziedzinie terapii genowych może prowadzić do wprowadzenia nowych, skuteczniejszych metod leczenia fenyloketonurii w przyszłości.
Należy jednak pamiętać, że terapie genowe wymagają dalszych badań i testów, zanim zostaną zatwierdzone do użytku klinicznego. Współpraca ze specjalistami oraz prowadzenie badań klinicznych są kluczowe w kontynuowaniu rozwoju terapii genowych dla fenyloketonurii.
IX. Wsparcie psychologiczne i edukacyjne
A. Znaczenie wsparcia dla rodzin
Wsparcie psychologiczne może pomóc rodzinom radzić sobie z emocjonalnymi aspektami PKU, takimi jak poczucie winy, stres czy frustracja związana z restrykcyjną dietą i innymi ograniczeniami. Może również poprawić komunikację w rodzinie i pomóc w budowaniu zdrowych relacji.
Wsparcie dla rodzin PKU może obejmować udział w grupach wsparcia, terapii rodzinnej, poradnictwa psychologicznego czy kontaktu z organizacjami pacjentów z PKU. Dźwiękowa się wsparcie i rada ze strony innych rodzin, które przeżywają podobne wyzwania, może być niezwykle cenne.
B. Strategie edukacyjne dla pacjentów z PKU
Pacjenci z PKU powinni być edukowani na temat wpływu diety na ich zdrowie i jak unikać żywności bogatej w fenylalaninę. Powinni również być informowani o konsekwencjach spożycia nadmiaru fenylalaniny oraz o znaczeniu regularnego monitorowania poziomu fenyloalaniny we krwi.
Wsparcie edukacyjne dla pacjentów z PKU może również obejmować udział w szkoleniach dotyczących przestrzegania diety, świadomości zdrowotnej i umiejętności radzenia sobie z sytuacjami, w których trudno uniknąć pokarmów bogatych w fenylalaninę.
Pacjenci powinni być również informowani o dostępnych zasobach i organizacjach, w których mogą znaleźć dodatkowe wsparcie, jak poradnictwo dietetyczne, poradnictwo genetyczne czy specjalistyczne kliniki zajmujące się fenyloketonurią.
X. Życie z Fenyloketonurią
A. Wyzwania w codziennym życiu

- Monitorowanie diety:
- Jednym z głównych wyzwań dla osób z fenyloketonurią jest konieczność ścisłego monitorowania diety. Pacjenci muszą regularnie kontrolować spożycie fenylalaniny, aby utrzymać jej poziom na bezpiecznym poziomie. To może wiązać się z liczeniem gramatury białka, unikaniem żywności bogatej w fenylalaninę oraz regularnym sprawdzaniem poziomu tego związku we krwi.
- Niedotrzymanie restrykcyjnej diety niskobiałkowej może prowadzić do wzrostu poziomu fenylalaniny we krwi, co z kolei może skutkować uszkodzeniem mózgu, opóźnieniem rozwoju, problemami z zachowaniem, a nawet niedorozwojem intelektualnym.
- Radzenie sobie z ograniczeniami społecznymi
- Pacjenci z fenyloketonurią często muszą radzić sobie z ograniczeniami społecznymi wynikającymi z restrykcyjnej diety. Mogą czuć presję społeczną związana z unikaniem pewnych potraw, niezrozumienie otoczenia czy utrudnienia w uczestnictwie w różnych wydarzeniach społecznych, gdzie dostępne są potrawy niezgodne z ich dietą.
B. Fenyloketonuria a życie zawodowe
- Wybór kariery
- Fenyloketonuria może wywrzeć wpływ na wybór zawodu, ponieważ niektóre ścieżki zawodowe mogą wiązać się z większymi trudnościami w zachowaniu diety niskobiałkowej. Pacjenci z PKU muszą brać pod uwagę swoje ograniczenia dietetyczne podczas wyboru kariery, aby móc skutecznie zarządzać swoim stanem zdrowia.
- Ważne jest, aby pacjenci z fenyloketonurią konsultowali się z specjalistami, aby uzyskać porady dotyczące wyboru kariery i jakie środki ostrożności należy podjąć, aby zapewnić sobie bezpieczne warunki pracy.
- Radzenie sobie w środowisku pracy
- Pacjenci z PKU mogą napotkać wyzwania w środowisku pracy związane z koniecznością dostosowywania diety do warunków pracy. Mogą też napotkać trudności w zakresie spełniania wymagań dietetycznych podczas podróży służbowych, spotkań biznesowych czy innych wydarzeń zawodowych.
- Współpraca z pracodawcą w zakresie zapewnienia odpowiednich warunków do przestrzegania diety oraz informowanie współpracowników o specyficznych potrzebach żywieniowych pacjenta z PKU może pomóc w radzeniu sobie z wyzwaniami w środowisku pracy.
C. Fenyloketonuria a zdrowie psychiczne
- Stres i lęk
- Osoby z fenyloketonurią mogą doświadczać stresu i lęku związanego z koniecznością ciągłego monitorowania diety, utrzymywania poziomu fenylalaniny we krwi, a także związanych z tym ograniczeń społecznych i zawodowych. Stres i lęk mogą mieć negatywny wpływ na ogólne samopoczucie i zdrowie psychiczne.
- Ważne jest, aby pacjenci z PKU szukali wsparcia psychologicznego i umieli radzić sobie ze stresem i lękiem poprzez techniki relaksacyjne, aktywność fizyczną, terapię czy inne metody redukujące napięcie.
- Radzenie sobie z depresją
- W niektórych przypadkach pacjenci z fenyloketonurią mogą zmagać się z depresją związaną z trudnościami związanymi ze stanem zdrowia, ograniczeniami diety, presją społeczną czy innymi czynnikami związanymi z chorobą. W czasie trudności emocjonalnych ważne jest, aby szukać pomocy i wsparcia.
- Terapia psychologiczna, wsparcie terapeutyczne oraz otwarta komunikacja z bliskimi mogą pomóc w radzeniu sobie z depresją i poprawie zdrowia psychicznego pacjenta z PKU. W razie potrzeby warto także skonsultować się z lekarzem specjalistą lub psychoterapeutą.
XI. Postępy w badaniach nad Fenyloketonurią
A. Nowe terapie i leki
W dziedzinie badań nad fenyloketonurią (PKU) dokonywane są postępy w zakresie poszukiwania nowych terapii i leków. Jednym z najbardziej obiecujących rozwiązań jest terapia zastępcza polegająca na podawaniu preparatu, który zawiera formę lizyny fenylalaniny (Phe) niezbędną dla organizmu, ale pozbawioną fenyloalaniny. Przykładowo, soprano, stosowany do leczenia PKU, jest preparatem zawierającym lizynę fenylalaninową.
B. Możliwości prewencji
Badania nad fenyloketonurią skupiają się również na poszukiwaniu możliwości prewencji tej choroby genetycznej. Jednym z podejść jest wprowadzenie testów przesiewowych, które umożliwią wczesne wykrycie PKU u noworodków. Dzięki wczesnej diagnozie możemy podjąć szybkie działania i zastosować odpowiednie leczenie, aby zmniejszyć ryzyko wystąpienia powikłań związanych z nadmiernym stężeniem fenyloalaniny we krwi.
XII. Edukacja i świadomość społeczna
A. Znaczenie kampanii edukacyjnych
Kampanie edukacyjne dotyczące fenyloketonurii są niezwykle istotne, ponieważ są przeznaczone zarówno dla pacjentów i ich rodzin, jak i dla społeczeństwa jako całości. Poprzez kampanie edukacyjne można przekazywać informacje na temat objawów, możliwych skutków oraz sposobów leczenia PKU. Dzięki temu zwiększa się świadomość społeczna na temat tej rzadkiej choroby genetycznej, co przyczynia się do eliminacji stereotypów i ułatwia integrację osób dotkniętych PKU w społeczeństwie.
B. Wsparcie dla rodzin i pacjentów
Wspieranie rodzin i pacjentów z fenyloketonurią odgrywa istotną rolę w procesie leczenia i przystosowania się do choroby. Dostęp do odpowiednich źródeł informacji, wsparcia emocjonalnego oraz finansowego może znacząco poprawić jakość życia osób z PKU. Organizacje pacjentów i grupy wsparcia odgrywają ważną rolę w zapewnianiu praktycznej pomocy oraz budowaniu społeczności osób zmagających się z fenyloketonurią. Ponadto, wsparcie ze strony specjalistów, takich jak dietetycy czy psychologowie, może pomóc pacjentom i ich rodzinom w radzeniu sobie z codziennymi wyzwaniami związanymi z PKU.

XIII. Podsumowanie
A. Kluczowe informacje o Fenyloketonurii
Fenyloketonuria (PKU) jest rzadką chorobą genetyczną, która prowadzi do zaburzeń metabolizmu aminokwasu fenyloalaniny. Osoby z PKU nie są w stanie prawidłowo przetwarzać fenyloalaniny, co prowadzi do jej gromadzenia się we krwi i mózgu. Bez odpowiedniego leczenia, wysokie poziomy fenyloalaniny mogą prowadzić do poważnych uszkodzeń mózgu i różnych objawów neurologicznych.
B. Najważniejsze aspekty choroby
PKU może mieć różne objawy, takie jak opóźnienie umysłowe, trudności w nauce, trudności w kontrolowaniu impulsów, problemy z zachowaniem i koncentracją. Wczesne rozpoznanie i wdrożenie odpowiedniej diety z ograniczeniem fenyloalaniny są kluczowe dla minimalizacji objawów i zapobiegania poważnym problemom zdrowotnym związanym z PKU.
C. Znaczenie wczesnej diagnozy i leczenia
Wczesna diagnoza PKU jest niezwykle ważna, ponieważ umożliwia podjęcie odpowiednich środków zaradczych i leczenie. Test przesiewowy na fenyloketonurię jest rutynowo przeprowadzany u noworodków w wielu krajach. W przypadku pozytywnego wyniku, leczenie powinno być rozpoczęte natychmiast w celu ograniczenia spożycia fenyloalaniny przez dietę i suplementację preparatami specjalnymi.
D. Znaczenie wsparcia społecznego i medycznego
Pacjenci z PKU i ich rodziny potrzebują wsparcia zarówno ze strony społeczności, jak i od specjalistów medycznych. Wsparcie emocjonalne, informacyjne i praktyczne jest niezbędne w procesie radzenia sobie z chorobą i jej skutkami. Specjaliści, takie jak dietetycy, psycholodzy i lekarze specjalizujący się w PKU, mogą dostarczać wartościowe informacje i wskazówki dotyczące prowadzenia odpowiedniej diety, monitorowania zdrowia i radzenia sobie z trudnościami na różnych etapach życia pacjenta z PKU. Wsparcie społeczne również odgrywa ważną rolę, umożliwiając pacjentom i ich rodzinom uczestnictwo w społeczeństwie i zmniejszając potencjalną marginalizację.
XIV. Najczęściej Zadawane Pytania (FAQ)
A. Czym jest fenyloketonuria i jak wpływa na organizm?
Fenyloketonuria (PKU) to rzadka choroba metaboliczna, spowodowana defektem enzymu odpowiedzialnego za przekształcanie fenyloalaniny. W wyniku tej dysfunkcji fenyloalanina gromadzi się we krwi i mózgu, co może prowadzić do uszkodzeń mózgu, opóźnienia umysłowego, trudności w nauce oraz innych objawów neurologicznych.
B. Jakie są możliwości leczenia PKU?
Głównym sposobem leczenia fenyloketonurii jest dieta niskobiałkowa, ograniczająca spożycie fenyloalaniny. Ponadto, pacjenci z PKU mogą stosować preparaty zawierające aminokwasy i specjalne formuły żywieniowe, aby zapewnić organizmowi niezbędne składniki odżywcze, jednocześnie ograniczając spożycie fenyloalaniny.
C. Czy możliwe jest całkowite wyleczenie fenyloketonurii?
Na obecnym etapie brak jest możliwości całkowitego wyleczenia fenyloketonurii. Jednak wczesna diagnoza i prowadzenie odpowiedniej diety mogą zminimalizować objawy choroby i zapobiec poważnym skutkom zdrowotnym związanym z nadmiarem fenyloalaniny.
D. Jakie są długoterminowe konsekwencje życia z PKU?
Osoby z fenyloketonurią mogą doświadczać różnych długoterminowych konsekwencji, takich jak trudności w nauce, problemy ze zdrowiem psychicznym, ewentualne uszkodzenia mózgu oraz ryzyko zaburzeń psychicznych. Regularne monitorowanie poziomu fenyloalaniny i ścisła dieta mogą pomóc w zarządzaniu tymi skutkami.
E. Jak wspierać osoby z fenyloketonurią w codziennym życiu?
Wspieranie osób z PKU w codziennym życiu wymaga zrozumienia ich specyficznych potrzeb dietetycznych oraz emocjonalnych. Ważne jest dostosowanie diety do ograniczeń fenyloalaniny, zapewnienie dostępu do specjalistycznej opieki medycznej i świadczenie wsparcia emocjonalnego. Edukacja społeczeństwa na temat fenyloketonurii oraz dostosowanie środowiska do specjalnych potrzeb pacjentów to również istotne elementy wspierania osób z tą chorobą.