
I. Wprowadzenie do Alkaptonurii
A. Krótka charakterystyka
Alkaptonuria to rzadka dziedziczna choroba metaboliczna, która jest spowodowana mutacją w genie HGD (homogentyzynaza). Ta mutacja powoduje, że organizm nie jest w stanie prawidłowo przetwarzać aminokwasów takich jak fenyloalanina i tyrozyna. W wyniku tego procesu enzymatycznego zaburzenia, substancja zwana homogentyzyną zostaje nieprawidłowo rozłożona, tworząc ciemnobrązowy pigment, który gromadzi się w różnych tkankach i narządach.
B. Historyczne tło


Alkaptonuria była jedną z pierwszych dziedzicznych chorób metabolicznych, która została opisana. W 1902 roku sir Archibald Garrod, brytyjski lekarz i naukowiec, zidentyfikował ją jako skuteczną chorobę metaboliczną. Był to przełomowy moment, ponieważ Garrod po raz pierwszy wykazał związek między genami a chorobami. Zauważył on, że pacjenci z alkaptonurią mieli ciemno zabarwioną mocz, co skłoniło go do zbadania tej choroby.
W dalszych badaniach Garrod wykazał, że choroba dziedziczy się w sposób autosomalny recesywny, co oznacza, że osoba musi odziedziczyć mutowany gen od obu rodziców, aby rozwinąć alkaptonurię.
Od tamtej pory badania nad alkaptonurią wykazały wiele informacji na temat skutków tej choroby. Niekontrolowane gromadzenie się ciemnego pigmentu w tkankach może prowadzić do poważnych problemów zdrowotnych, takich jak uszkodzenie stawów, serca i nerek. Badania naukowe nad alkaptonurią nadal trwają, w nadziei na rozwinięcie nowych metod leczenia i lepsze zrozumienie tego rzadkiego schorzenia.
II. Genetyka Alkaptonurii
A. Wzorce dziedziczenia
Alkaptonuria jest dziedziczona w sposób autosomalny recesywny. Oznacza to, że potrzeba dwóch kopii mutowanego genu HGD, jednej od matki i drugiej od ojca, aby osoba rozwijała chorobę. Osoby, które mają tylko jedną kopię mutowanego genu, są nosicielami choroby i zazwyczaj nie wykazują objawów. Jeśli obydwoje rodzice są nosicielami alkaptonurii, istnieje 25% szansa, że ich dziecko odziedziczy dwie kopie mutowanego genu i rozwinie chorobę.
B. Mutacje genetyczne
Alkaptonuria jest spowodowana mutacją w genie HGD, który jest odpowiedzialny za produkcję enzymu zwanej homogentyzynazą. Ta mutacja powoduje nieprawidłowe funkcjonowanie enzymu, co prowadzi do niezdolności organizmu do prawidłowego przetwarzania aminokwasów, takich jak fenyloalanina i tyrozyna. W wyniku tej mutacji enzym nie jest w stanie skutecznie rozłożyć homogentyzyny, co prowadzi do jej akumulacji w organizmie.
Mutacje w genie HGD mogą występować na różnych poziomach, od pojedynczych zmian w kodonach do większych delecji lub insercji. Te mutacje wpływają na funkcję enzymu homogentyzynazy, co prowadzi do akumulacji homogentyzyny oraz powstawania ciemnobrązowego pigmentu w tkankach.
Rozpoznanie mutacji genetycznych w alkaptonurii jest ważne dla postawienia diagnozy i oceny ryzyka dziedziczenia choroby. Badania genetyczne mogą być wykorzystane do identyfikacji mutacji w genie HGD, co pomaga ustalić, czy dana osoba ma alkaptonurię, jest nosicielem lub jest podatna na jej rozwoju.
III. Biochemiczna Podstawa Alkaptonurii
A. Zaangażowane szlaki metaboliczne
Alkaptonuria jest spowodowana defektem metabolicznym, który wpływa na przemiany aminokwasów fenyloalaniny i tyrozyny. Zaangażowane szlaki metaboliczne w alkaptonurii to:
- Szlak metabolizmu fenyloalaniny: Fenyloalanina jest aminokwasem, który jest przekształcany w tyrozynę przez enzym fenylalaninę hydroksylazę. U osób z alkaptonurią enzym ten nie działa prawidłowo, co prowadzi do braku przekształcenia fenyloalaniny w tyrozynę.
- Szlak metabolizmu tyrozyny: Tyrozyna jest kolejnym aminokwasem, który jest przekształcany w kwas homogentyzynowy przez enzym hydrolazę homogentyzynową. W alkaptonurii enzym ten również jest defektywny, co prowadzi do gromadzenia się kwasu homogentyzynowego.
B. Akumulacja kwasu homogentyzynowego
Mutacja w genie HGD powoduje, że enzym hydrolaza homogentyzynowa nie działa prawidłowo. W wyniku tego nieprawidłowego funkcjonowania enzymu kwas homogentyzynowy nie jest prawidłowo metabolizowany i ulega akumulacji w organizmie. Ten kwas ma zdolność do tworzenia ciemnobrązowego pigmentu, który odkłada się w różnych tkankach i narządach, w tym w skórze, stawach, tkance łącznej, sercu i nerkach.
Akumulacja kwasu homogentyzynowego i powstawanie pigmentu jest główną przyczyną objawów klinicznych alkaptonurii, takich jak ciemno zabarwiony mocz, ciemne plamy na skórze i uszkodzenie tkanki łącznej. Te objawy mogą prowadzić do różnych problemów zdrowotnych, takich jak zapalenie stawów, kamienie nerkowe, zesztywnienie kręgosłupa i uszkodzenia serca.
Zrozumienie biochemicznej podstawy alkaptonurii jest kluczowe dla rozwoju strategii leczenia i terapii, które mogą pomóc w redukcji akumulacji kwasu homogentyzynowego oraz minimalizacji objawów i powikłań związanych z chorobą.
IV. Objawy Kliniczne
A. Zaangażowanie stawów
Alkaptonuria powoduje powstawanie pewnych objawów klinicznych, w tym zapalenie stawów, które jest wynikiem akumulacji kwasu homogentyzynowego w tkankach stawowych. Ten proces prowadzi do uszkodzenia chrząstki stawowej i zwiększonego ryzyka osteoartrozy. Są to objawy głównie występujące u dorosłych z alkaptonurią i częściej dotyczą stawów kręgosłupa, bioder, kolan i barków.
B. Ochronosis: Zmiany barwnikowe skóry i tkanek
Alkaptonuria jest również związana z odkładaniem się ciemnych plam na skórze i tkanek. Te obszary z odkładającym się pigmentem są nazywane ochronosisem. Chociaż plamy charakterystyczne dla ochronosisu są nieszkodliwe, mogą one być kosmetycznym i psychologicznym problemem dla niektórych chorych. Ochronosis może dotyczyć różnych tkanek, w tym skóry, chrząstki stawowej, mięśni i serca.
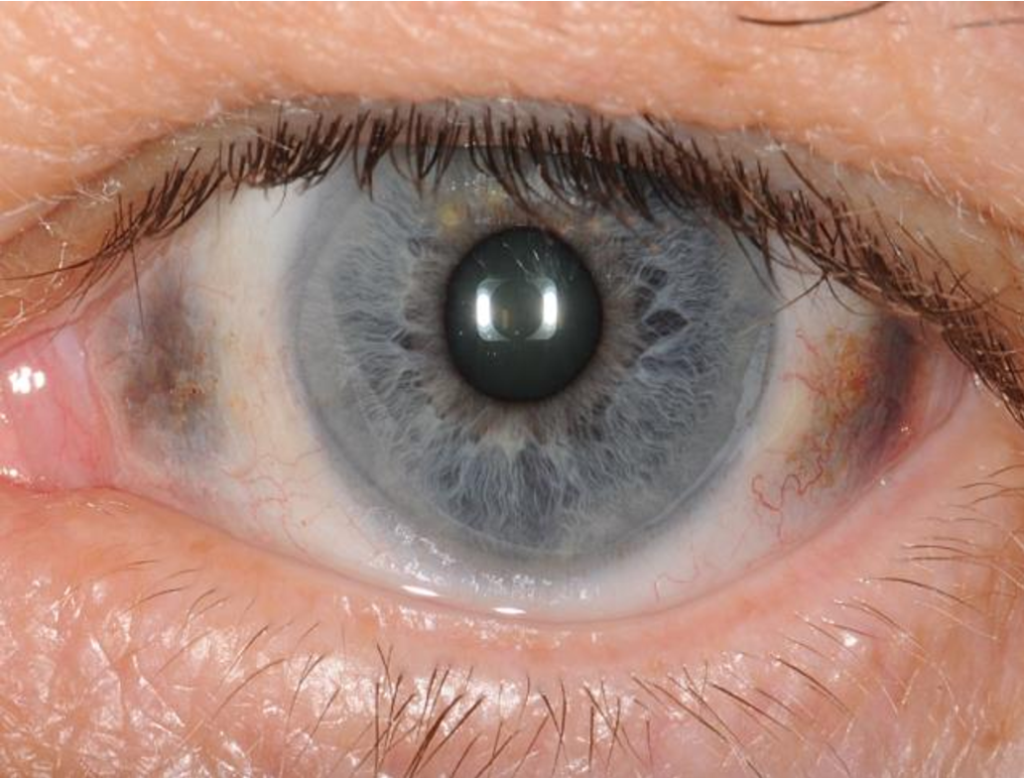
Inne objawy kliniczne, które mogą występować w alkaptonurii, to kamienie nerkowe, zespół oczopląsu, jaskra, zespół przysadkowy i inne problemy zdrowotne. Objawy te są bardziej zróżnicowane wśród pacjentów z alkaptonurią i mogą różnić się w zależności od stopnia akumulacji kwasu homogentyzynowego i miejsca, w którym jest on odkładany.
Znając objawy kliniczne alkaptonurii, lekarze mogą lepiej diagnozować i leczyć tę chorobę. Wraz z postępem w terapii genowej i medycynie personalizowanej naukowcy będą kontynuować pracę nad identyfikacją najlepszych strategii leczenia i terapii dla pacjentów z alkaptonurią.
V. Diagnoza Alkaptonurii
A. Badanie kliniczne
Podczas badania klinicznego lekarz przeprowadza szczegółowy wywiad medyczny i fizykalne badanie pacjenta. Lekarz może zwrócić uwagę na charakterystyczne objawy i cechy kliniczne, które sugerują występowanie alkaptonurii.

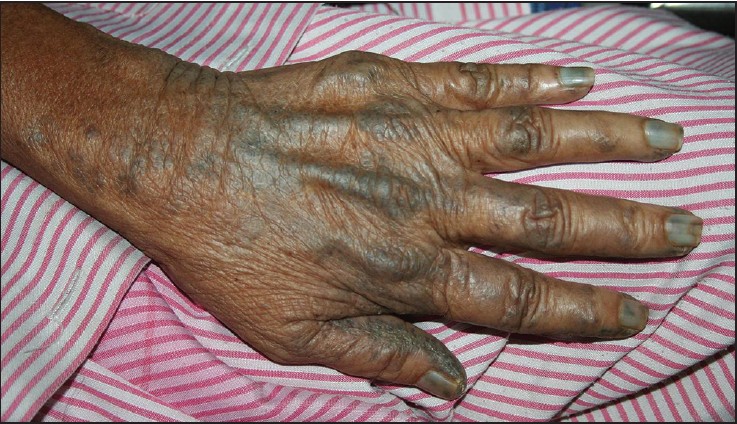
- Objawy skórne: Chorzy na alkaptonurię często mają ciemne plamy na skórze, zwłaszcza na małżowinach usznych, nosie, dłoniach, okolicy narządów płciowych i wokół oczu. Te plamy nazywane są ochronosisem. U niektórych pacjentów ochronosis może także obejmować inne obszary, takie jak sutek czy przestrzeń międzykostna. Ochronosis może prowadzić do pogrubienia skóry, tworzenia guzków lub owrzodzeń.
- Objawy stawowe: Alkaptonuria jest związana z uszkodzeniem stawów, które objawia się sztywnością i bólem stawów. Częściej dotyczy stawów kręgosłupa, bioder, kolan i barków. Akumulacja kwasu homogentyzynowego w tkance stawowej prowadzi do przyspieszonego zużycia chrząstki stawowej i ryzyka wystąpienia osteoartrozy.
B. Testy laboratoryjne
Testy laboratoryjne są kluczowe w diagnozowaniu alkaptonurii i potwierdzaniu obecności akumulacji kwasu homogentyzynowego.
- Badanie moczu: Analiza moczu jest podstawowym testem laboratoryjnym. Mocz pacjentów z alkaptonurią jest ciemnobrązowy lub czarny, co wynika z obecności kwasu homogentyzynowego. Test moczu może również wykryć obecność homogentyzyny i produktów jej przemiany, takich jak kwasy fenoloksyetylo- i fenylosyrenowy.
- Badanie poziomu aminokwasów we krwi: W alkaptonurii może występować podwyższony poziom fenyloalaniny we krwi, ze względu na defekt enzymu fenylalaniny hydroksylazy. Stwierdzenie podwyższonego poziomu fenyloalaniny może sugerować możliwość alkaptonurii.
- Badanie genetyczne: Badanie genetyczne może być również wykonane w celu potwierdzenia diagnozy alkaptonurii. Analiza mutacji w genie HGD, który jest odpowiedzialny za alkaptonurię, może dostarczyć pewności co do obecności choroby u pacjenta.
W przypadku podejrzenia alkaptonurii, odpowiednie badania laboratoryjne i diagnostyczne są kluczowe dla ustalenia właściwej diagnozy. Wczesna diagnoza i zarządzanie alkaptonurią mogą pomóc w minimalizowaniu objawów i powikłań związanych z tą rzadką chorobą genetyczną.
VI. Diagnoza Różnicowa
A. Rozróżnianie Alkaptonurii od podobnych schorzeń
- Ochronosis bez Alkaptonurii:
- Ochronosis jest stanem, w którym dochodzi do osadzania się czarnych pigmentów w tkankach, w tym w chrząstkach stawowych. Może on być spowodowany przez czynniki zewnętrzne, takie jak długotrwałe stosowanie leków zawierających składniki chemiczne, na przykład hydrochinon. Ochronosis bez alkaptonurii nie jest związany z akumulacją kwasu homogentyzynowego, który jest charakterystyczny dla alkaptonurii. W odróżnieniu od alkaptonurii, ochronosis nie wiąże się z ciemnym zabarwieniem moczu.
- Czarna skóra związana z innymi przyczynami:
- Ciemne zabarwienie skóry może występować w innych schorzeniach, takich jak hemochromatoza lub zatrucie ołowiem, które mogą powodować nagromadzenie substancji chemicznych w organizmie. Hemochromatoza, zaburzenie genetyczne charakteryzujące się nadmiernym gromadzeniem żelaza w organizmie, może również prowadzić do zabarwienia skóry na brązowoczarne. Z kolei zatrucie ołowiem może powodować szaro-czarną zmianę skórną.
- Artroza:
- Objawy stawowe, takie jak ból i sztywność stawów, które występują u pacjentów z alkaptonurią, mogą być podobne do objawów osteoartrozy (artrozy). Osteoartroza jest chorobą zwyrodnieniową stawów, która dotyka głównie starsze osoby. Istotne jest zwrócenie uwagi na różnice w składzie moczu oraz na charakterystyczne objawy skórne, które mogą pomóc odróżnić alkaptonurię od osteoartrozy.
- Zespół Dennyego-Drummonda:
- Zespół Dennyego-Drummonda to rzadkie schorzenie genetyczne charakteryzujące się zaburzeniami metabolizmu aminokwasów, w tym fenyloketonurią. Pacjenci z tym zespołem mogą mieć podwyższony poziom fenyloalaniny we krwi, podobnie jak pacjenci z alkaptonurią. Konieczne jest przeprowadzenie dokładnych badań laboratoryjnych, w tym analizy moczu i poziomu aminokwasów we krwi, aby postawić właściwą diagnozę różnicową.
W przypadku podejrzenia alkaptonurii, istotne jest przeprowadzenie dokładnej diagnozy różnicowej, aby wykluczyć inne schorzenia, które mogą mieć podobne objawy kliniczne. Współpraca z zespołem medycznym oraz przeprowadzenie odpowiednich badań laboratoryjnych są kluczowe dla postawienia właściwej diagnozy oraz wdrożenia odpowiedniego leczenia.
VII. Metody Leczenia
A. Zarządzanie objawowe
Nie ma specyficznej terapii dla alkaptonurii, ale strategie zarządzania objawowego mogą pomóc w minimalizowaniu skutków choroby. Należą do nich:
- Leczenie farmakologiczne bólu: Leki przeciwbólowe mogą być wymagane, aby pomoc w kontrolowaniu bólu wynikającego z uszkodzenia stawów.
- Leczenie chirurgiczne: Chirurgia może być wymagana w przypadku znacznego uszkodzenia stawów, zwykle w celu wymiany stawów.
- Terapia rehabilitacyjna: Fizjoterapia i trening siłowy mogą pomóc w zwiększeniu ruchomości stawów oraz wzmocnieniu mięśni.
B. Nowe terapie
Badania nad terapiami zaradczymi dla alkaptonurii prowadzone są w wielu krajach. Wpisują się w nie m.in.:
- Terapia genowa: Terapia genowa to eksperymentalna metoda leczenia polegająca na wprowadzeniu zdrowego genu do komórek pacjenta, w celu naprawienia defektu genetycznego. W przypadku alkaptonurii, wprowadzenie zdrowego genu do komórek pacjenta może pomóc w zapobieganiu akumulacji kwasu homogentyzynowego.
- Terapia enzymatyczna: Terapia enzymatyczna opiera się na wprowadzeniu aktywnego enzymu HGD do organizmu pacjenta, którego brak prowadzi do alkaptonurii. Eksperymenty przeprowadzane na modelach zwierzęcych pokazały, że metoda może zmniejszyć ilość kwasu homogentyzynowego w organizmie.
- Terapia małymi cząsteczkami: Jest to eksperymentalna terapia, która opiera się na stosowaniu leków, których celem jest hamowanie akumulacji kwasu homogentyzynowego w organizmie lub inhibitorem HGD. Obecnie trwają badania nad kwasem asparaginowym i inhibitory HGD jako potencjalnych metod leczenia alkaptonurii.
Wprowadzenie nowych terapii leczniczych do praktyki klinicznej może zająć wiele lat. Na razie strategie zarządzania objawowego są nadal podstawowym podejściem do leczenia pacjentów z alkaptonurią. Jednak rozwój medycyny personalizowanej oraz badań nad lekami i terapiami genowymi daje nadzieję na poprawienie jakości życia osób cierpiących na tę chorobę.
VIII. Wpływ na Jakość Życia
A. Implikacje fizyczne
- Uszkodzenie stawów: Alkaptonuria może prowadzić do uszkodzenia stawów, co może powodować ból i sztywność. W zaawansowanych przypadkach może być konieczne przeprowadzenie operacji wymiany stawów w celu przywrócenia funkcji stawów.
- Problemy z nerkami: Zwiększone ilości kwasu homogentyzynowego w organizmie mogą prowadzić do powstawania kamieni nerkowych, co z kolei naraża pacjentów na wystąpienie infekcji dróg moczowych oraz inne powikłania związane z układem moczowym.
- Zaburzenia układu sercowo-naczyniowego: Niektóre badania sugerują, że alkaptonuria może zwiększać ryzyko wystąpienia pewnych schorzeń sercowo-naczyniowych, takich jak miażdżyca.
B. Wyzwania psychospołeczne
- Depresja i lęk: Pacjenci z alkaptonurią mogą doświadczać depresji i lęku związanych z chronicznymi dolegliwościami, bólem, ograniczeniami w codziennych czynnościach i prognozą choroby. Wsparcie psychologiczne i terapia mogą być ważne w radzeniu sobie z tymi wyzwaniami.
- Ograniczenia w codziennym życiu: Alkaptonuria może wpływać na zdolność pacjentów do wykonywania codziennych czynności, takich jak chodzenie, praca czy dbanie o siebie. Ograniczenia te mogą wpływać na jakość życia pacjentów i wymagać adaptacji i wsparcia.
- Społeczne wykluczenie: Choroba rzadka jak alkaptonuria może prowadzić do społecznego wykluczenia pacjentów. Zrozumienie choroby i edukacja społeczeństwa na jej temat może pomóc w zmniejszeniu stygmatyzacji i poprawie jakości życia pacjentów.


Pacjenci z alkaptonurią często muszą stawić czoła wielu fizycznym oraz psychospołecznym wyzwaniom, które mogą znacząco wpływać na ich jakość życia. Wsparcie medyczne, rehabilitacja, terapia psychologiczna oraz edukacja są istotne w zapewnieniu kompleksowej opieki i poprawieniu zdrowia psychicznego i fizycznego pacjentów z alkaptonurią.
IX. Badania i Postępy
A. Obecne badania
- Badania nad mechanizmami choroby: Badacze prowadzą badania mające na celu zrozumienie dokładnych mechanizmów, przez które alkaptonuria prowadzi do uszkodzenia stawów, nerek i innych narządów. To zrozumienie może przyczynić się do opracowania bardziej skutecznych strategii leczenia.
- Badania nad lekami i terapiami zaradczymi: Trwają badania nad nowymi lekami i terapiami zaradczymi dla alkaptonurii, takimi jak terapia genowa, terapia enzymatyczna i terapia małymi cząsteczkami. Te eksperymentalne terapie mają na celu zmniejszenie akumulacji kwasu homogentyzynowego w organizmie i zapobieganie konsekwencjom choroby.
- Badania nad diagnostyką: Badacze pracują nad opracowaniem bardziej skutecznych metod diagnostycznych alkaptonurii, takich jak testy genetyczne i biochemiczne. To pozwoli na szybszą i bardziej precyzyjną diagnozę oraz wcześniejsze rozpoczęcie terapii.
B. Kierunki przyszłych badań
- Badanie długoterminowych efektów terapii: Warto kontynuować badania nad długoterminowymi efektami terapii zaradczych dla alkaptonurii, aby ocenić ich skuteczność i bezpieczeństwo oraz zrozumieć, jakie korzyści i ograniczenia mogą wynikać z ich stosowania na dłuższą metę.
- Badania nad lepszymi strategiami zarządzania bólem: Ból jest jednym z głównych objawów alkaptonurii, dlatego ważne jest kontynuowanie badań nad lepszymi strategiami zarządzania bólem u pacjentów z tą chorobą. Poszukiwanie nowych leków przeciwbólowych oraz terapii niefarmakologicznych może pomóc poprawić jakość życia pacjentów.
- Badania nad poprawą jakości życia: Warto prowadzić badania nad sposobami poprawy jakości życia pacjentów z alkaptonurią. Dotyczy to zarówno aspektów fizycznych, jak rehabilitacja i terapia fizyczna, jak i aspektów psychospołecznych, takich jak terapia psychologiczna i działania mające na celu zmniejszenie społecznego wykluczenia.
Kontynuacja badań nad alkaptonurią i postęp w dziedzinie nauk medycznych daje nadzieję na rozwinięcie nowych i bardziej efektywnych strategii zarządzania tą chorobą. Badania te mają na celu zwiększenie długości i jakości życia pacjentów oraz zmniejszenie negatywnego wpływu choroby na ich codzienne funkcjonowanie.
X. Środki zapobiegawcze
A. Poradnictwo genetyczne
Poradnictwo genetyczne odgrywa kluczową rolę w przypadku alkaptonurii, ponieważ jest to dziedziczna choroba genetyczna. Oto kilka aspektów poradnictwa genetycznego związanych z alkaptonurią:
- Diagnoza przedpoborowa – Poradnictwo genetyczne może pomóc rodzinom, które mają historię alkaptonurii, w określeniu, czy mają ryzyko dziecka z chorobą. Przy pomocy testów genetycznych można zidentyfikować mutacje genetyczne związane z alkaptonurią u rodziców i stwierdzić, czy mają one szansę na przekazanie choroby potomstwu.
- Prognozowanie ryzyka – Poradnictwo genetyczne może pomóc w zrozumieniu ryzyka dziedziczenia alkaptonurii przez potencjalne potomstwo. Genetyk może omówić z pacjentem lub rodziną prawdopodobieństwo dziedziczenia mutacji genetycznej oraz możliwości testów prenatalnych, które pozwalają na wczesne wykrycie alkaptonurii u nienarodzonego dziecka.
- Wsparcie emocjonalne – Poradnictwo genetyczne oferuje również wsparcie emocjonalne dla osób i rodzin dotkniętych alkaptonurią. Dostarcza informacji i narzędzi, które pozwalają na bardziej świadomą i lepszą opiekę nad osobami z tą rzadką chorobą.
B. Zalecenia dotyczące stylu życia
Zarządzanie alkaptonurią wymaga również odpowiednich modyfikacji stylu życia, aby zmniejszyć objawy i poprawić jakość życia. Oto kilka zaleceń dotyczących stylu życia dla osób z alkaptonurią:
- Regularne badania kontrolne – Osoby z alkaptonurią powinny regularnie odwiedzać lekarza i poddawać się badaniom kontrolnym w celu monitorowania stanu zdrowia i oceny postępu choroby. Badania obejmują między innymi badania krwi, monitorowanie funkcji nerek i stawów oraz testy obrazowe takie jak rezonans magnetyczny.
- Dieta z ograniczeniem tyrozyny i fenyloalaniny – Osoby z alkaptonurią powinny unikać pokarmów bogatych w tyrozynę i fenylalaninę, które są prekursorami kwasu homogentyzynowego. Dieta bogata w owoce, warzywa, białko roślinne i niskotyrozynową formę białka zwierzęcego może pomóc w zmniejszeniu ilości kwasu homogentyzynowego w organizmie.
- Aktywność fizyczna i rehabilitacja – Regularna aktywność fizyczna, w tym ćwiczenia odpowiadające możliwościom pacjenta, może pomóc w utrzymaniu sprawności stawów i opóźnieniu postępu choroby. Fizjoterapia oraz ćwiczenia wzmacniające i rozciągające mogą również zmniejszyć ból stawów i poprawić ruchomość.
- Wsparcie psychologiczne – Życie z alkaptonurią może wiązać się z wieloma wyzwaniami emocjonalnymi. Dlatego ważne jest, aby osoby dotknięte tą chorobą otrzymywały wsparcie psychologiczne. Terapia indywidualna lub grupowa mogą być pomocne w radzeniu sobie z lękiem, depresją i innymi trudnościami emocjonalnymi spowodowanymi chorobą.
Ważne jest, aby osoby z alkaptonurią przedyskutowały z lekarzem i specjalistami poradnictwa genetycznego swoje indywidualne potrzeby dotyczące zarządzania chorobą i stosowania zaleceń dotyczących stylu życia.
XI. Alkaptonuria a Ciąża
A Rozważania dla przyszłych matek
Kiedy kobieta z alkaptonurią planuje ciążę, istnieje kilka kwestii, które warto omówić z lekarzem i specjalistą ds. poradnictwa genetycznego. Oto kilka ważnych rozważań dla przyszłych matek z alkaptonurią:
- Dieta i suplementacja – Kobieta powinna omówić z lekarzem dietę, która minimalizuje spożycie tyrozyny i fenyloalaniny, aby zmniejszyć produkcję kwasu homogentyzynowego. Mogą również być zalecane suplementy, takie jak witamina C, które mogą pomóc w ograniczeniu odkładania się pigmentu w tkankach.
- Monitorowanie stanu zdrowia – Podczas ciąży ważne jest monitorowanie stanu zdrowia kobiety i płodu. Kobieta z alkaptonurią powinna regularnie odwiedzać lekarza i poddawać się badaniom kontrolnym, aby monitorować stany stawów, nerek i innych organów.
- Ryzyko dziedziczenia – Kobieta z alkaptonurią powinna skonsultować się z lekarzem i specjalistą ds. poradnictwa genetycznego, aby omówić ryzyko dziedziczenia choroby przez dziecko. Można omówić możliwości testów prenatalnych i wczesnego wykrywania alkaptonurii u nienarodzonego dziecka.
- Wsparcie emocjonalne – Ciąża może być zarówno ekscytującym, jak i stresującym czasem dla przyszłych matek. Kobieta z alkaptonurią może potrzebować wsparcia emocjonalnego, aby poradzić sobie z lękami i obawami związanymi z chorobą i ciążą. Ważne jest, aby szukać wsparcia od bliskich, specjalistów ds. poradnictwa genetycznego, a także grup wsparcia dla matek z rzadkimi chorobami.
B. Rozważania dla przyszłych ojców
Również przyszli ojcowie z alkaptonurią powinni omówić niektóre kwestie z lekarzem i specjalistą ds. poradnictwa genetycznego przed planowaną ciążą. Oto kilka rozważań dla przyszłych ojców z alkaptonurią:
- Ryzyko dziedziczenia – Przyszły ojciec powinien skonsultować się z lekarzem i specjalistą ds. poradnictwa genetycznego w celu omówienia ryzyka dziedziczenia choroby przez potomstwo. Testy genetyczne mogą być przeprowadzone, aby ustalić, czy przyszły ojciec ma mutację genetyczną związaną z alkaptonurią.
- Wsparcie emocjonalne i efektywna komunikacja – Przyszły ojciec może przeżywać różne emocje i obawy związane z chorobą i roli ojca. Ważne jest, aby poszukiwać wsparcia emocjonalnego, zarówno u bliskich, jak i od specjalistów ds. poradnictwa genetycznego. Efektywne komunikowanie się z partnerką i otwarte rozmowy o obawach mogą również pomóc w zarządzaniu stresem i emocjami.
- Monitorowanie stanu zdrowia – Przyszły ojciec powinien również poddawać się badaniom kontrolnym i monitorować stan zdrowia, zwłaszcza w zakresie stawów i nerek. Kontrola regularnych badań może pomóc w zidentyfikowaniu ewentualnych zmian i objawów choroby.
Ważne jest, aby zarówno przyszła matka, jak i przyszły ojciec z alkaptonurią miały wsparcie medyczne i emocjonalne przed i w trakcie ciąży. Specjaliści ds. poradnictwa genetycznego i lekarze mogą zapewnić niezbędne informacje, wsparcie i opiekę, aby przejść przez ten okres bezpiecznie i zminimalizować ryzyko dla potomstwa.
XII. Grupy Wsparcia i Aktywizacji
A. Nawiązywanie kontaktu z społecznością
Jednym z ważnych aspektów opieki nad osobami z alkaptonurią jest nawiązywanie kontaktu z społecznością osób z tą chorobą. Grupy wsparcia i aktywizacji mogą stanowić cenne źródło wsparcia emocjonalnego, informacji, porad, oraz przeżycia związanego z chorobą.
- Wsparcie emocjonalne – Nawiązanie kontaktu z innymi osobami z alkaptonurią może pomóc w poczuciu wspólnoty i zrozumieniu, że nie jest się samym w walce z chorobą. W takiej grupie można znaleźć wsparcie emocjonalne i porady dotyczące radzenia sobie z różnymi aspektami choroby, takimi jak objawy, leczenie, dieta, i adaptacja do codziennego życia z alkaptonurią.
- Informacje i zasoby – Grupy wsparcia mogą być źródłem cennych informacji na temat najnowszych badań i postępów w dziedzinie leczenia alkaptonurii. Członkowie grup mogą dzielić się swoimi doświadczeniami z różnymi terapiami, suplementami i dietą, co może być pomocne dla innych osób dotkniętych chorobą.
- Poradnictwo i edukacja – W grupach wsparcia często spotykają się eksperci z dziedziny medycyny, dietetyki i rehabilitacji, którzy mogą udzielić porad i edukacji na temat zarządzania alkaptonurią. Takie spotkania mogą być również okazją do zadawania pytań i uzyskiwania odpowiedzi od profesjonalistów w dziedzinie alkaptonurii.
- Aktywizacja społeczna – Grupy wsparcia mogą organizować różnorodne aktywności społeczne, takie jak seminaria, konferencje, warsztaty i spotkania towarzyskie. Takie działania mogą pomóc w zwiększeniu świadomości społecznej na temat alkaptonurii oraz umożliwić osobom dotkniętym chorobą większe zaangażowanie społeczne i włączenie się w działania na rzecz poprawy opieki nad alkaptonurią.
- Wsparcie dla rodzin – Nie tylko osoby z alkaptonurią, ale również ich rodziny mogą znaleźć wsparcie w grupach wsparcia. Rodzice dzieci z alkaptonurią mogą dzielić się swoimi doświadczeniami i radami dotyczącymi opieki nad dzieckiem. Partnerzy i bliscy mogą również potrzebować wsparcia i zrozumienia w radzeniu sobie z chorobą i jej wpływem na codzienne życie.
XIII. Adresowanie Mitów i Błędnych Przekonań
A Powszechne nieporozumienia
Niestety, Alkaptonuria jest mało rozpoznawalną chorobą, co prowadzi do występowania wielu mitów i błędnych przekonań. Ważne jest, aby odpowiednio edukować społeczeństwo i rozwiewać powszechne nieporozumienia na temat Alkaptonurii. Oto kilka powszechnych nieporozumień i jak im przeciwdziałać:
- Alkaptonuria to tylko problem z kolorami moczu – To jedno z największych nieporozumień dotyczących tej choroby. Alkaptonuria to rzadka genetyczna choroba metaboliczna, która prowadzi do gromadzenia się toksycznego związku chemicznego w organizmie o nazwie homogentyzyny. Powoduje to szereg objawów, takich jak sztywność i bóle stawów, zaburzenia sercowo-naczyniowe, problemy z oddychaniem, zaburzenia wzroku, problemy z nerkami i inne.
- Alkaptonuria to jedynie problem kosmetyczny – Pomimo że wygląd moczu jest charakterystycznym objawem Alkaptonurii, nie jest to jedyny problem. Pacjenci z Alkaptonurią często doświadczają trudności z poruszaniem się z powodu bólu stawów i sztywności. Mogą także mieć problemy z sercem, oczami, nerkami i innymi układami ciała.
- Choroba jest zaraźliwa – Alkaptonuria jest dziedziczną chorobą genetyczną i nie jest zaraźliwa. Nie można jej złapać od innej osoby poprzez kontakt fizyczny.
- Nie ma leczenia dla Alkaptonurii – Chociaż nie ma skutecznego leczenia Alkaptonurii, istnieją pewne strategie zarządzania, które mogą pomóc w łagodzeniu objawów i wpływie choroby na codzienne życie. To może obejmować regularne badania, fizykoterapię, odpowiednią dietę oraz leczenie objawowe.
- Osoby z Alkaptonurią mają skróconą długość życia – Pomimo że Alkaptonuria może prowadzić do wielu poważnych problemów zdrowotnych, tacy jak choroby serca, osoby z tą chorobą zazwyczaj żyją tak długo, jak osoby bez Alkaptonurii, o ile ich objawy są odpowiednio zarządzane i leczone.
XIV. Wpływ na Dynamikę Rodzinną
A. Strategie radzenia sobie
Alkaptonuria może mieć znaczący wpływ na dynamikę rodziną, ponieważ choroba wymaga ciągłej opieki i może wpływać na codzienne życie i funkcjonowanie rodziny. W związku z tym, istnieją strategie radzenia sobie, które mogą pomóc w zarządzaniu tym wyzwaniem. Oto kilka przykładów:
- Komunikacja – Ważne jest, aby członkowie rodziny otwarcie rozmawiali o chorobie, jej wpływie na codzienne życie i potrzebach osoby z alkaptonurią. Zapewnienie otwartości i szczerą komunikację pomaga zrozumieć i zaspokoić potrzeby każdego członka rodziny.
- Edukacja – Dobra znajomość alkaptonurii i jej objawów może pomóc członkom rodziny w lepszym zrozumieniu choroby i podejmowaniu odpowiednich działań. Ważne jest, aby wszyscy byli świadomi ograniczeń, jakie może stwarzać alkaptonuria i jak można zapewnić odpowiednią opiekę i wsparcie.
- Wspólne poszukiwanie wsparcia – Rodzina może znaleźć wsparcie w grupach wsparcia dla osób z alkaptonurią oraz w innych organizacjach zajmujących się tą chorobą. Można również skorzystać z poradnictwa rodzinnego, aby uzyskać wsparcie i przewodnictwo w zarządzaniu chorobą jako rodzina.
- Ustalanie granic i przystosowanie – Alkaptonuria może wymagać pewnych modyfikacji w stylu życia rodziny, takich jak dostosowanie diet, zwiększenie czasu na fizykoterapię czy inne zabiegi medyczne. Ważne jest, aby wszyscy członkowie rodziny byli świadomi i zaangażowani w te zmiany.
- Opieka i wsparcie emocjonalne – Alkaptonuria może być trudnym doświadczeniem zarówno dla osoby dotkniętej chorobą, jak i dla innych członków rodziny. Ważne jest, aby zapewnić sobie nawzajem opiekę i wsparcie emocjonalne. To może obejmować rozmowy o uczuciach, udział w grupach wsparcia lub terapii rodzinnej.
XV. Edukacja Personelu Medycznego
A. Wzmacnianie świadomości i wiedzy
Wzmacnianie świadomości i wiedzy personelu medycznego na temat Alkaptonurii jest kluczowe, aby zapewnić właściwą diagnostykę, leczenie i opiekę dla pacjentów dotkniętych tą chorobą. Oto kilka punktów, które należy rozwinąć podczas edukacji personelu medycznego:
- Przyczyny i objawy – Personel medyczny powinien mieć świadomość, że Alkaptonuria jest genetyczną chorobą metaboliczną, która powoduje gromadzenie się toksycznego związku chemicznego o nazwie homogentyzyna. Powinni być w stanie rozpoznać charakterystyczne objawy Alkaptonurii, takie jak ból stawów, sztywność, zaburzenia sercowo-naczyniowe, zaburzenia wzroku i problemy z nerkami.
- Metody diagnostyki – Personel medyczny powinien być dobrze zaznajomiony z metodami diagnostycznymi Alkaptonurii, takimi jak badanie moczu, które można przeprowadzić w celu sprawdzenia obecności homogentyzyny. Lekarze powinni znać procedury diagnostyczne i umieć zinterpretować wyniki.
- Zarządzanie objawami i leczenie – Personel medyczny powinien być świadomy różnych sposobów zarządzania objawami Alkaptonurii i dostępnych metod leczenia. Mogą obejmować odpowiednią rehabilitację, fizykoterapię, farmakoterapię lub nawet interwencję chirurgiczną w niektórych przypadkach. Wiedza o tych możliwościach pomoże w odpowiednim leczeniu pacjentów.
- Śledzenie pacjentów – Wzmacnianie świadomości personelu medycznego na temat potrzeby regularnego monitorowania pacjentów z Alkaptonurią jest kluczowe. Osoby z Alkaptonurią powinny regularnie poddawać się badaniom, takim jak badania laboratoryjne, badania wizualne czy diagnostyka obrazowa. Dzięki temu można śledzić postęp choroby i skuteczność leczenia.
- Wsparcie pacjenta i porady genetyczne – Personel medyczny powinien być w stanie zapewnić wsparcie emocjonalne pacjentom związane z Alkaptonurią i pomóc im zrozumieć aspekty genetyczne choroby. Porady genetyczne przyszłym rodzicom i rodzinom są również ważne dla zrozumienia dziedziczenia Alkaptonurii i podejmowania odpowiednich decyzji.
Wzmacnianie świadomości i wiedzy personelu medycznego na temat Alkaptonurii jest istotne dla poprawy jakości opieki dla pacjentów z tą rzadką chorobą. Kontynuacja edukacji poprzez szkolenia, materiały edukacyjne i współpracę z organizacjami zajmującymi się Alkaptonurią pomoże w lepszym zrozumieniu i radzeniu sobie z tym schorzeniem.
XVI. Podsumowanie Kluczowych Punktów
A. Podsumowanie istotnych informacji
Podsumowanie kluczowych punktów dotyczących Alkaptonurii pomaga ułatwić zrozumienie tej rzadkiej choroby i podsumować jej istotne informacje. Oto kilka punktów, które można uwzględnić w podsumowaniu:
- Definicja i przyczyny – Alkaptonuria to rzadka dziedziczna choroba metaboliczna, która powoduje gromadzenie się toksycznego związku chemicznego o nazwie homogentyzyna. Jest spowodowana mutacją w genie HGD, który jest odpowiedzialny za produkcję enzymu zwalczającego homogentyzynę.
- Objawy – Najważniejszym objawem Alkaptonurii jest ciemnienie moczu po utlenieniu, co jest spowodowane obecnością homogentyzyny. Inne objawy mogą obejmować ból i sztywność stawów, problemy z sercem, wzrokiem i układem moczowym.
- Diagnostyka – Diagnoza Alkaptonurii może być stwierdzana poprzez badanie moczu w celu wykrycia homogentyzyny. Wynik pozytywny może być potwierdzony przez badania genetyczne w celu identyfikacji mutacji w genie HGD.
- Postępowanie – Alkaptonurię można zarządzać poprzez łagodzenie objawów i komplikacji choroby. W przypadku bólu stawów i sztywności może być stosowana rehabilitacja, fizjoterapia i leki przeciwbólowe. Niektóre pacjenci mogą wymagać interwencji chirurgicznej w celu poprawy funkcji stawów.
- Wsparcie psychologiczne i porady genetyczne – Pacjenci z Alkaptonurią często wymagają wsparcia emocjonalnego ze względu na wpływ choroby na ich codzienne życie. Porady genetyczne są również ważne, zarówno dla pacjentów, jak i dla ich rodzin, aby zrozumieć dziedziczenie choroby i podjąć odpowiednie decyzje w planowaniu rodziny.
XVII. Najczęściej Zadawane Pytania (FAQ)
A. Odpowiedzi na powszechne pytania
Odpowiedzi na najczęściej zadawane pytania dotyczące Alkaptonurii są istotne dla podniesienia świadomości i zrozumienia tej rzadkiej choroby. Oto kilka powszechnych pytań i odpowiedzi związanych z Alkaptonurią:
- Co to jest Alkaptonuria?
Alkaptonuria to rzadka choroba metaboliczna, w której organizm nie jest w stanie prawidłowo rozkładać homogentyzyny – toksycznego związku chemicznego. Skutkuje to gromadzeniem się homogentyzyny w tkankach i narządach, co może prowadzić do różnych objawów i powikłań. - Jakie są objawy Alkaptonurii?
Głównym objawem Alkaptonurii jest ciemnienie moczu po utlenieniu. Inne częste objawy to ból i sztywność stawów, problemy z sercem, wzrokiem i układem moczowym. Objawy mogą występować od dzieciństwa lub młodości, jednak niektóre osoby mogą nie prezentować żadnych objawów przez długi czas. - Czy Alkaptonuria jest dziedziczna?
Tak, Alkaptonuria jest dziedziczna. Jest spowodowana mutacją w genie HGD, który jest odpowiedzialny za produkcję enzymu, który metabolizuje homogentyzynę. Dlatego choroba może być przekazywana z pokolenia na pokolenie. - Jak diagnozować Alkaptonurię?
Diagnoza Alkaptonurii zwykle opiera się na badaniu moczu w celu wykrycia obecności homogentyzyny. Dodatkowo, potwierdzenie diagnozy może wymagać analizy genetycznej w celu identyfikacji mutacji w genie HGD. - Czy istnieje leczenie dla Alkaptonurii?
Obecnie nie ma lekarstwa na Alkaptonurię. Zarządzanie chorobą skupia się na łagodzeniu objawów i komplikacji. Może obejmować fizjoterapię, leki przeciwbólowe, a w niektórych przypadkach, interwencję chirurgiczną. - Czy Alkaptonuria wpływa na długość życia?
Osoby z Alkaptonurią mogą mieć różne przebiegi choroby. Większość pacjentów prowadzi normalne życie, ale może doświadczać trudności związanych z objawami. W niektórych przypadkach, przewlekłe powikłania Alkaptonurii mogą wpływać na długość życia. - Czy można zapobiec Alkaptonurii?
Ponieważ Alkaptonuria jest chorobą genetyczną, jej zapobieganie polega na świadomości dziedziczenia choroby. Osoby z historią choroby w rodzinie mogą skonsultować się z doradcą genetycznym przed planowaniem rodziny w celu zrozumienia ryzyka i podjęcia odpowiednich decyzji.